You know...sometimes I see papers with huge titles like this and think....oh no....what are we overselling today....
This is a study that deserves a big big big title.
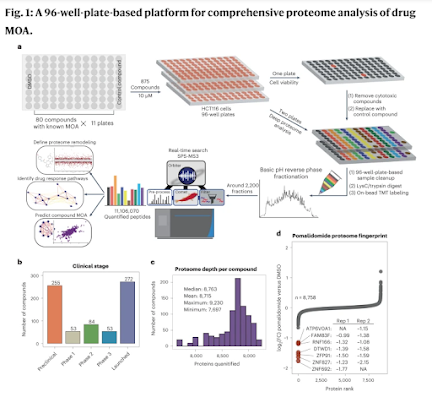
This relatively small group of authors did proteomics on 875 drugs.
Eight hundred and seventy five.
achthundertfünfundsiebzig
八百七十五
שמונה מאות שבעים וחמש
åttahundrasjuttiofem
ottehundrede og femoghalvfjerds
walóng daán at pitóng pû’t limá
ثمانية مائة وخمسة وسبعون
ochocientos setenta y cinco
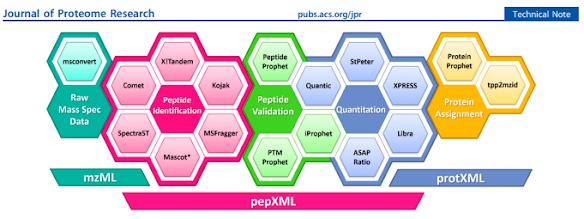
How'd they do it? This figure pretty much covers it!
Okay, so who cares, right? I'm not going to download 11 million spectra and neither is anyone in the Pharmacology department I work in.
To make it useful, they'd have to have a ridiculously easy web interface that allows you to look at any drug you want and how it affects around 9,000 different proteins.
Welcome to
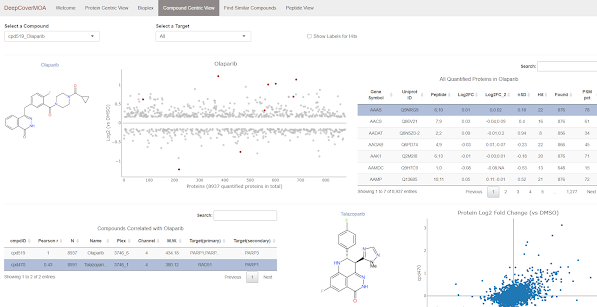
Check this out. I know someone who studies/develops compounds similar to Olaparib. I know every drug has like 12 different synonyms, but -- I'll type the first 3 letters into the compounds box and BOOOM!
Not only can I pick a specific protein to see if it is perturbed by Olaparib treatment, but this resource goes way further than that. It generates correlation plots between it and the other drugs in the library based on the protein level effects!
This is so so so so so so so so so good. I'm just floored by how much thought went into this. There are, of course, similar things for drug + mRNA levels that everyone uses (most of the biggest ones are old microarray data...blech....) but people use those all the time. Protein level??? Nothing I've ever seen has ever come close.