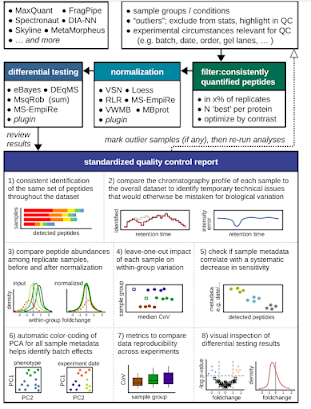
Yikes! Okay, y'all. I'm seeing way way too many DIA experiments without any kind of contaminant libraries being used. Not throwing out any names, but this goes all the way to spectral libraries included in something commercial I paid a lot for.
Recent work really went into how very important this is even if the title was a little tone deaf.
This isn't hard to do, at all, and I know one increasingly grumpy old guy who might be a complete jerk during peer review it you aren't doing it.
If you are doing things like "library free" this is super easy. If you aren't it is also easy.
Library free for PROSIT or DIA-NN or whatever?
Just append your FASTA database you are using for prediction to have contaminants.
If you don't have NotePad++ on your PC, you should. I'm pretty sure that Bruker instruments come with it preinstalled on the instrument PCs now. It is free, it doesn't append silly things to the name of your file and it appears to be able to open documents of almost limitless size.
Open your FASTA you were going to predict or whatever and open your favorite contaminants libary. Cut/Paste them together. You can go with the classic: cRAP (
https://www.thegpm.org/crap/) or you can download the MaxQuant contaminant library.
Very related: Charlotte Dawson has this great discussion on contaminant libraries as part of the campProtR package they developed (Charlotte and Tom Smith @ Cambridge?) as well as direct links to download various contaminant FASTAs. Totally worth skimming through and I'm definitely checking out that R tool.
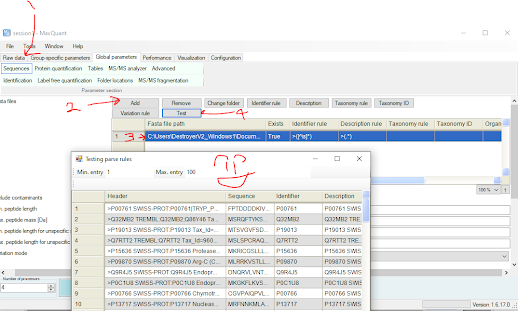
Looking at the contaminant library I have above, I don't think that these annotations are going to look perfect in everything. I'm appending in 12 different KRAS mutants from SwissProt and they don't have colons or semicolons. They use |, so I'm going to use some quick Ctrl+R, Replace All, starting with : for | so they all look the same. Once it looks like they'll pass (you can always proof read your FASTA with the free tool in the PD viewer or in MaxQuant to verify you don't have a bunch of mistakes in your FASTA (or probably 75 other tools).
5 steps for MaxQuant below. Chances are if MaxQuant can parse your FASTA properly now you're good to go
Merge your FASTA. I'm just going to go to the shorter one, right click on the screen, Select All, Cut, and paste it at the bottom. Then I'm going to save mine as HumanUniProt04162010_12KRAS121322_contaminants.fasta (part of that is a joke).
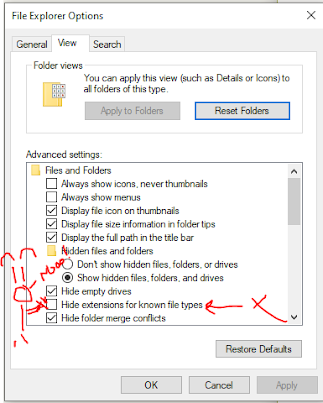
This is on here somewhere, but Windows has this default in most cases that you may need to turn off --
You can search for Folder options or file explorer options. If this is enabled when you save a file .fasta it will save it as .fasta.txt just to mess with you.
Now you've got a FASTA! If I'm going to use PROSIT, I use the EncylopeDIA to make the input file. Here is the walkthrough for that.
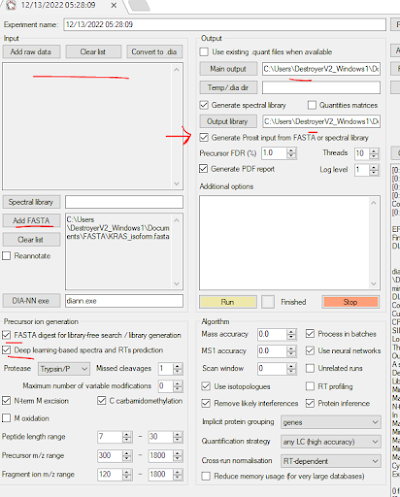
Something I only learned recently was that you don't actually have to process data in DIA-NN. You can just use it to make you spectral libraries. It will also generate your Prosit input if you have DIA-NN but you don't have EncyclopeDIA (which I highly recommend you have, it's amazing).
Here I've just had it take my FASTA -- no input files and generate my spectral library and Prosit input. You can also do something funny with DIA-NN where you give it one spectral library format and it will give you it's favorite but I should go to work soon.
Either way -- here you should now have a predicted spectral library with contaminant in it.
Want to generate a library with one? Chances are (I hope!) you already have! If you are searching your DDA data with a good contaminant library to make your input -- don't filter them out before making your spectral library from your data. That's it. I know a lot of tools or templates autofilter out the ++ contaminants or whatever. Remove those filters before building your library. I build my libraries using Skyline and then convert them to whatever format I need with EncyclopeDIA.
Hopefully you don't need any of this information and you're like "geez, Ben, great way to waste 38 minutes of your life (I actually type sort of slow)".