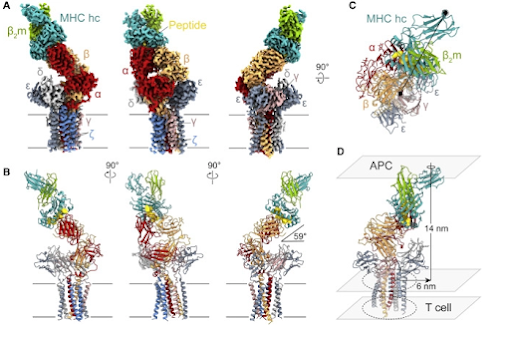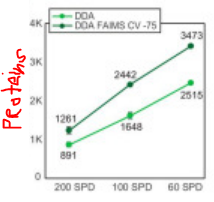
What do we know so far? A little.
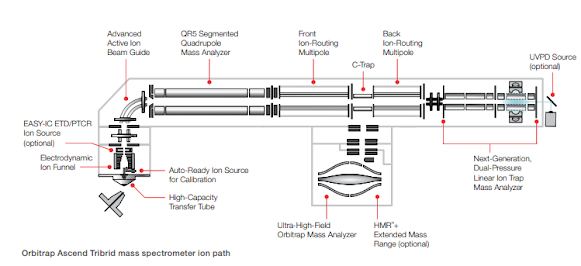
The schematics appear to show two separate ion routing multipoles, which can only increase parallelization capabilities. Oh cool! A full document with schematics is up on the website link above now!
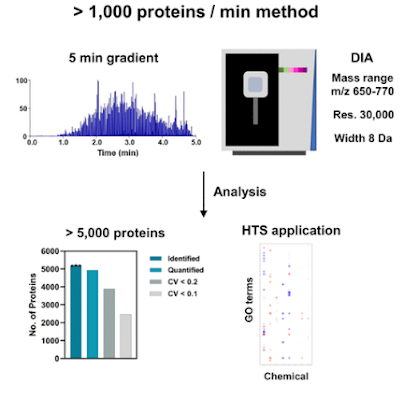
Some pretty amazing numbers here. The optional HighMassRange option goes to 16,000 m/z. That might be the highest number outside of the E+EMR or UHMR (I forget that latter's upper number). Orbitrap is talking about 45 Hz @ 7,500 resolution?
The autoready ion source appears to be a self-calibrating option, which would explain some of the marketing stuff we're seeing cool videos of. The document says you can auto schedule calibrations in your queue which is an Orbitrap first.
For anyone hoping that we'd see a TOF back there, you might be disappointed, but it looks like the ion trap was again tinkered with to improve UVPD, and PTCR.
It seems like full details will release at IMSC which just wasn't on my radar this year and I don't know where I had that cognitive gap to not even consider putting it on the calendar. For the first big launch from this vendor in quite a long time, and a really impressively professional series of marketing hints and links, I suspect it will be quite the official launch. Sad I'll miss it, but hopefully some data will leak and more will show up at that little conference in rural Mexico in December.