I just stumbled on this while working on a tough deadline or three, and now I can cite it! It is a preprint from a couple of years ago.
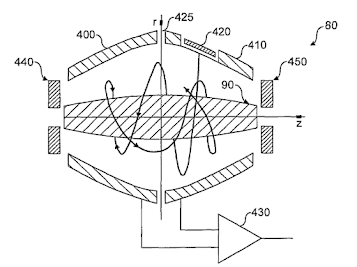
Due to the time of flight (effect, not instrument) coming out of the quadrupole on the TIMSTOF you can't really scan low and high mass fragment ions simultaneously. They take longer to get there and you didn't get fined by the union that "maintains" your building for altering your ceiling tiles because you wanted to scan slooooow. (BTW: I'm 100% pro-union, labor unions are the only thing between lots of members of my family dying in coal mines in West Virginia so people like the...governor....of the state can save $10. He is really really really rich, btw, largely by operating coal mines unsafely and owes millions of dollars in safety violations that he just doesn't pay because billionaires in the US can literally do anything they want with absolutely no repercussions. I have personally known people who have died underground in mines owned by that walking pile of dogshit, though, thankfully no family, so please pardon the emotion here.)
What was I....oh yeah! This preprint!

Okay, so don't quote me on this preprint at all. The data isn't publicly available anywhere and it's been sitting on a preprint server a couple of years, but I think the concept is cool.
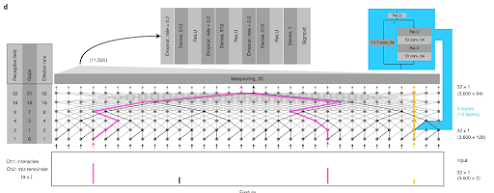
I was looking to see if anyone who has shared with me how they are doing glycoproteomics on these ceiling disrupting monsters has published their approach so I can cite it. Why I'm interested is that I really fine tune out my pre-pulse storage conditions for my two TMT scan events and crank the collision energy to 11 on the scan that liberates my reporter ions.
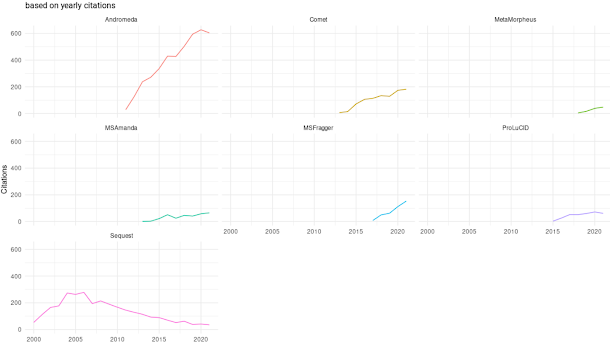
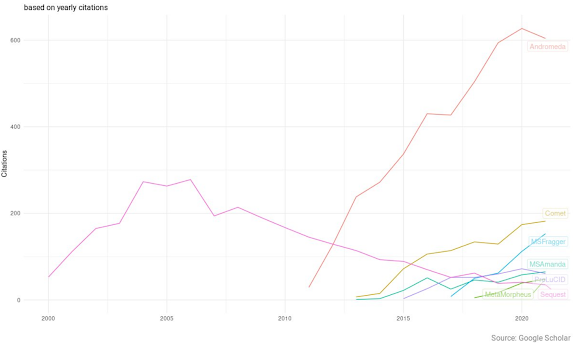
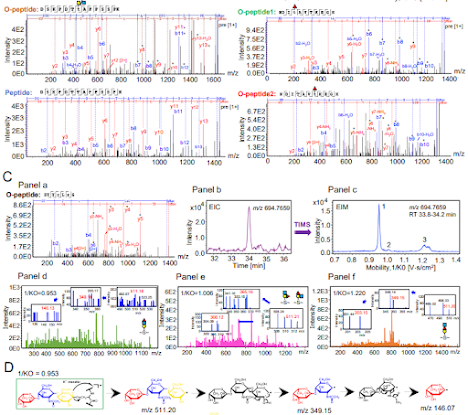
The end result is that in my MS2 spectra I often see crazy high intensity reporter ions and then a great big gap up to around 230 or so before we see the peptide signal. (They look like the spectra at the very top -- see where the signal starts? Ain't gonna see that 204 or 183. It looks like there is nothing in the 204 range. Which causes a lot of the glycoproteomics programs to output nothing at all because a lot of them only consider spectra for glycan ID if they see oxonium ions. This group uses PEAKS and they just don't think about oxonium ions. Boom. Problem solved, maybe?