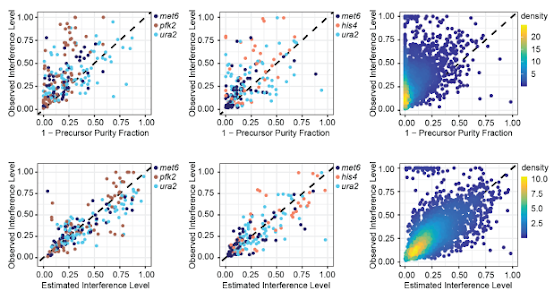
One thing that we've got to think about is the dreaded zero value, or the Interference Free Index! I'm using the methods described by Paulo et al., and those are awesome. Though...worth noting, you used to be able to upload any LCMS data into their toolkit, but now it has to be from one specific vendor. I had to build my own. That will be in the tutorial if I ever finish it.
The goal of the IFI is to understand how much background you're acquiring and when you've got a ground truth like "this yeast DOES NOT EXPRESS" this protein, that's a darned nice zero point.
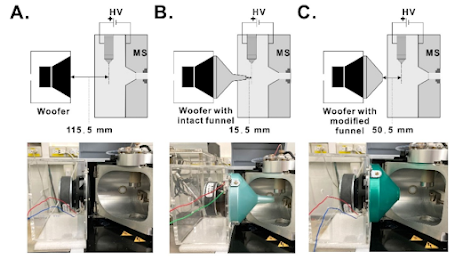
As you might expect, MS3 based approaches for reporter ion quan outperform anything MS2 I've seen, though really tuning in your chromatography and high resolution ion mobility can have a dramatic effect.
But -- if you understand your zero point ground truth fully (aka, zero looks like this number) and you have some values that you absolutely know the quantification on, can you do something with that?
Could you, for example, build a causal model? And use that to readjust your quantification?
Wooooo.....that's some grown up math......and probably not for me. I'd rather just draw an arbitrary line, let's go with...I dunno....2-fold and then just send that out to people!
These nerds, however, decided to see what they could do with the grown up math.

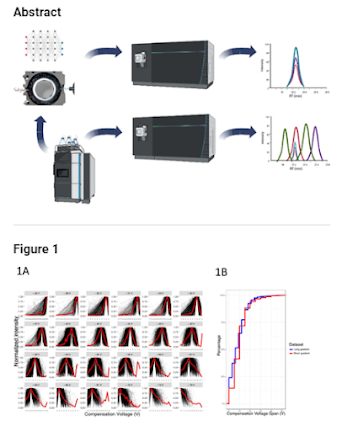
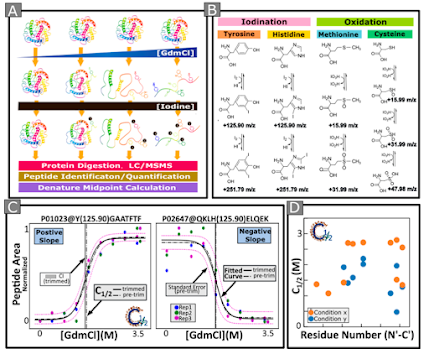
They took knowns standards and modeled it out (using FAIMS + MS2 based quan), then built out that statistical model to differentiate their known values.
Guess what -- it looks AWESOME, and it almost completely decompresses the dreaded reporter ion quan ratio compression.
This isn't the first attempt at something like this, but it may very well be the simplest. Could I reproduce this? Not today, I got up at 3am and I'm still super behind. I skipped breakfast and lunch so I could type this. But this group has a bad habit of making their tools freely and publicly available, so maybe I don't even have to try. I guess we'll see!