This is on the list of papers I've been looking forward to reading since USHUPO 2021!
For some reason, people actually sometimes care about more than the linear protein sequence. They care about what it looks like before you break all the bonds and cover it in SDS and cut it into little pieces. I did this study section thing I can't tell you about, mostly because the sleep deprivation it required left me unable to form long term memories. Based on what I can read of my notes I'm sorry to say NMR and baseball still appear to be a things that people do or go watch or however that works and the flashiness of CryoEM is definitely able to distract people from really nice LCMS proteomics work.
Chalf (See-Half) is part of a pipeline that looks really complicated at first, but is really elegant and suprisingly simple to set up.
The best part is that you don't have to do 1 purified protein at a time (I bet those 6-10 histidines at the end don't actually alter those folding or melting dynamics anyway..... ) With this, you can do ALL the proteins. Want proof? This team did HUMAN PLASMA.

Sounds impossible, right?
You remember guanidine? For some reason it was a component of some soft drinks in the 1990s, and the rumor was that it was derived from bat poo. One of the reasons that the 90s was the greatest decade in the history of mankind was that our population density had just gotten to a point where urban legends were passed around at their peak level and access to ways to check whether there actually was bat poo in that can of soda were very limited. Maybe that information was on the message boards somewhere? Who knows?
Guanidine resurfaced in mass spectrometry for a while as a way to linearize proteins! Focus on that because I think the soft drink might have been something different. GuanOdine? I don't know and I'm not going to look it up.
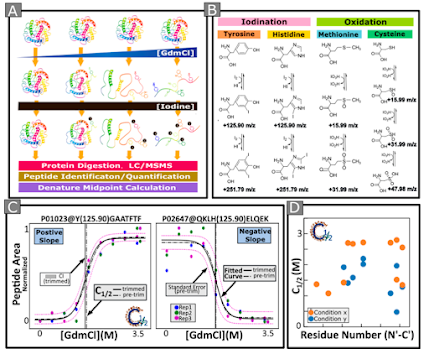
Cool part. Iodine apparently can't get in to react with amino acids if they're inside a 3D protein structure. It only works right if that amino acid is exposed to the liquid surface interface thingamabob.
As you add increasing amounts of guanidine proteins unfold to expose amino acids to iodine reaction as a direct function of their structural stability. RIGHT?!?!?!
Okay, but how would you process that data? Now you've got iodines on everything? It is actually more complicated than that, unfortunately. The free Iodine doesn't just bind to amino acids. Well, it does for some, adding two I to tyrosine and histidine, but it does other weird stuff to other amino acids.
The modified amino acid masses are all here in the paper, and they processed the data with PEAKs for both label free and TMT labeled samples (quan!). You don't have to do the whole structural stability caclulations yourself (thank you and whoever did the experiment that discovered you get extra energy from bat poo!) because you can get the software that does that here.
And that is actually what C-half or Chalf is. The workflow is called -I-P-S-A-, but that is the exact acronym for something else I use just about every day that I can only find by searching my blog for the link to it and I didn't want to make it difficult for me to find.
Hi, Ben!! I felt flattered that there are people actually waiting to read CHalf paper, moreover, think it is worth to spend of your time to write a blog for it. Thank you! We should do a better job on searching before we decided on the acronym XD
ReplyDelete