
How do these packages differ in some of the most important points we care about?
How do these packages differ in some of the most important points we care about?
Sebastian took pity on me and my attempts to draw straight lines on IMS windows and made these illustrations. Check out how sick this is.



I checked my horoscope this morning and it said that Houston ASMS is going to be probably the biggest year for high end mass spectrometry releases in history.
I quote from the passage for Capricorn:
Expect at least 3 vendors to release new flagship instruments that will push the boundaries of what you can get from the tiniest amounts of protein imaginable. If you purchased the most expensive LCMS instrument available in December of 2022 at list price, like the wise leaders of a weird little place called Janelia, and still have not hired anyone to run it, you might look even sillier than you do now.
Then I looked it up and realized that I'm actually a Cancer, but it was too late, I'd already typed this. Who knew that horoscopes were still around?
Disclaimers are over there -->
I have no currently valid NDAs and no one tells me anything, this is purely for comedy purposes for a joke about 8 people will get.
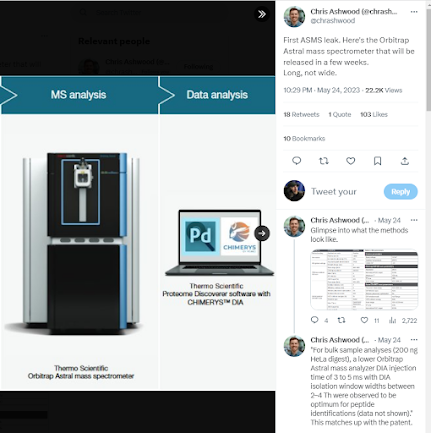
Whether intentional or not (US deadline for S10 high end shared equipment awards is next week, so...) a popular scientific media outlet for chemists has posted a couple of application notes for what appears to be the long-sought Orbi-TOF system.
Neither application note actually has details on the second mass analyzer (such as whether it is a TOF) but considering that the application note for plasma
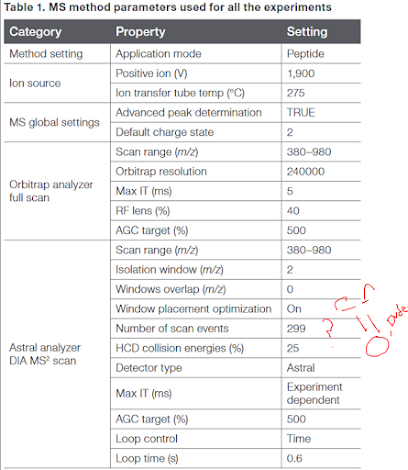
That's either an ion trap at a resolution of 14 at 200 m/z or 4 ion traps or a respectable TOF. Or....an Orbi-Orbi where your cycle time is 9 seconds or something, but look at the "Loop Time"!! That's 299 scan events in 0.6 seconds? And there is some ridiculously short chromatography in that app note.
A second app note goes down to 50 picograms cancer cell digest on column and kicks back something like 2,000 proteins.
You might notice there are no links here. I was recently in court as an expert witness and I really truly don't want to be on the other side of things. I'm dying to talk about that case which was an absolute microcosm of everything that is wrong with the current state of mass spectrometry, like why we aren't taken seriously as a distinct scientific field and why so many mass spec labs fail, but I gotta wait out some stuff.
You'll also notice that they made the great decision of extending the Assend nomenclature. I suspect they'll do something like Hyundai did with the new Genesis brand and we're going to keep going up. Betcha we're just a few years from Assgard. You can find this info yourself by googling the name of the instrument.

(Everyone was sort of on the edges -- and this was Ron Schnaar talking about super complex surface glycans (eeeek)-- the fact anyone is visible is a big thing in my mind! )
I started my first real job in clinical chemistry at Johns Hopkins 20 years ago this August (eeeeeeek!) and I've been bumming around the city, hospital and university off and on ever since. For all of that time, there really hasn't been much mass spectrometry here. If you total the hospital system and the university (which are separate entitities) there are just about 50,000 full time employees and 23,000 full time students. That's sort of big for a school that has never really had even a Metabolomics core.
SO...the fact that there were two rooms full of posters with a lot of people I DIDN'T EVEN KNOW showing all sorts of mass spectrometry data is super exciting.
Highlights for this reader base?
Chan-Hyun Na showed some great stuff on how his group is finding non-canonical peptides in Alzheimer's models. That is, of course, a protein disease that genomics isn't all that helpful with. While I'm a huge fan of the Steen lab work on this topic, I had no idea they were doing this super effectively across the street.
Stephen Fried doesn't just do really mass spectrometry to solve fundamental questions about the earliest stages of enzyme evolution (cool recent paper here). He demonstrated some applications of similar approaches to understand neurological decline.
Rahul Bharadwaj showed some work with Bob Cole's core (I think Lauren DeVine will have a poster at ASMS showing this data, it was too popular for me to get to it), super ridiculously small laser capture microdissection samples + high plex TMT proteomics (MS3 based quan, I think, I can't always read my notes) from people who were so amazing that they left their brains to the Lieber brain banks. Super cool stuff.
Ron Schnaar studies the most frustrating looking cell surface glycans of profound medical importance through photoreactive probes his team synthesizes themselves. He made a really solid argument for why intricate biological pull-down studies benefit from the super high accuracy TMT multiplexing you get on the Orbitraps largely designed for that purpose. When your biology and the molecular aspects of your experiment are super complex, it is nice to have the best quan on the back end that you can get (and the internal QC aspects of multiplexing).
Josh Smith talked about his continued work with adductomics -- there it is -- I was sure I had a blog post on a paper of his from a few years ago. If you aren't familiar, it is cool and scary stuff. Actually, that paper was on targeting -- here is a very recent JASMS study doing it untargeted!
That was only half the day! There were great posters (Bob O'Meally is making custom SureQuant panels, check out his poster if you're in Houston, it's really cool) and the rest of the day was imaging and them small molecules and there was even a single cell ICP-MS (accurately measuring metal concentrations in one cell at a time). Super cool day.
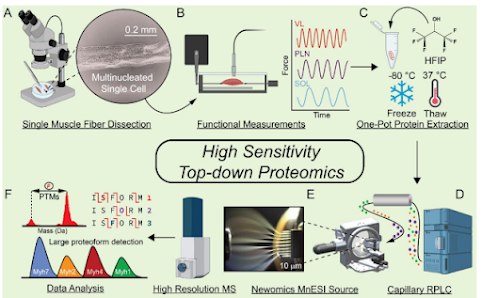
Woooo!! While most people are trying to get usable shotgun data out of their first actual single cell right now, there are a couple groups out there trying to do the impossible -- TOP DOWN proteomics from single cells.
During the Proteomics Show Road to Chicago we talked to both Alexander Ivanov and Ying Ge and I was surprised to find out they were both doing it -- and both have published now!
You can check out the new study from Ge lab here! There is an earlier preprinted version if you can't figure out how to log onto PNAS through your library....


I recently had a tirade about making these new fangled DIA data processing software runs faster. It was focused on SpectroNaut, but I think it will carry over to other tools equally.
Honestly, it was a desperation thing for me because we were bottlenecked like crazy at the data processing things and -- things don't seem nearly as desperate now.
These numbers this morning are the most striking to me.
The first thing SpectroNaut does is convert your whatever it is data type to the HTRMS data that it likes.
Check out how long it takes to convert 55,000 diaPASEF MS2 spectra from one file on a Samsung Pro SATA SSD drive


When you've got multiple grant proposals due first week of June and your front office wants them turned in Wednesday, saving 3 minutes each on 90-ish files on just the very first step of your processing pipeline can seem like a dogsend.



For some context from the mass spec side, if you have to get out an Orbitrap Eclipse and run a 2 hour gradient at nanoflow rates (IonOpticks 25cm!) using the ion trap so you have the highest sensitivity on your MS2s and then put on a FAIMS source to get a couple dozen peptides I'll go out on a limb and say -- there ain't much there. You have to be talking about a tiny tiny fraction of contamination, right?
Interesting to think about, and I'm cautiously optimistic that this is actually saying very positive things about the mAB production industry. However, I guess if that mAB came from a hamster ovary cell and I was deathly allergic to hamsters rather than just grossed out by them, I'd probably want to know those 60 peptides were there?
Put a question mark in the title in case this is something like part per Trillion detection and it isn't remotely biologically relevant and is just annoying to people making antibody drugs.
WOOOOOOOO!!! I'm leaving this here, then checking my calendar and seeing if I can install this without help. And then getting help. Or....doing whatever my calendar tells me to, then doing the things above.

Wait. I don't even have to install it to try it out.
Check out the PipeR shiny.io interface here!
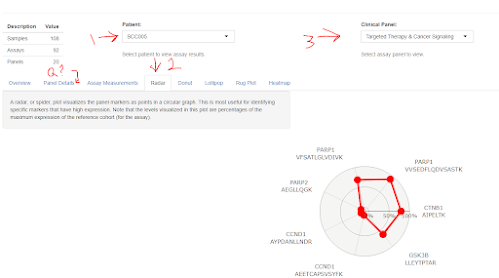
Wait. What was that panel again? BOOM! Click the panel details tab! It's like these people have called a sleepy resident at 3 am to deliver a crazy blood potassium level and this poor person who has been on the floor for 28 straight hours -- is like "remind me what the normal range is again, please...?" If you're the clinical chemist who has never had to pull more than a 16 hr shift on a holiday it's easy to be sort of dismissive of the physician, but there are reasons most people (even in the US) aren't allowed to work more than 16 hour shifts. You don't get smarter at hour 17. Reminders are embedded in reports that go out for very good reasons.
I can't over express how much I love this whole idea!
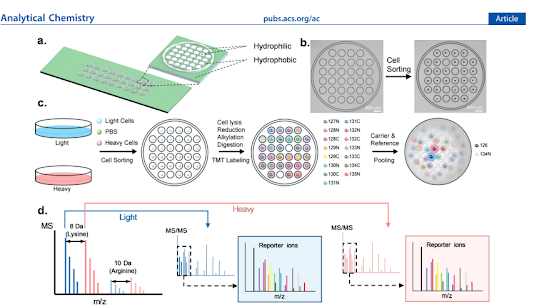
...wooo...and this is another way to get the throughput up!

My first obvious concern is -- OMG, the data processing must be miserable -- but as someone who is commonly combining hundreds of multiplexed TMT with randomized samples....this is a point where I've got zero room to talk.
They do see advantages from the carrier channels in terms of # of differential proteins between conditions, but the numbers are impressive in both cases. For those of us who aren't reducing and alkylating prior to TMT labeling single cells, the methods here for adding those steps are a welcome surprise.

It took about 7 months of doing diaPASEF every day, plus attending multiple talks (including two really great ones last week by Vadim Demichev and Matt Willetts, respectively) before I felt confident finishing this post. And I'm largely doing it to straighten out my own thoughts before I dive into altering the provided methods on my TIMSTOF SCP (which I strongly believe can be improved but it is such a miserable pain in the butt to generate super high quality actual single human cell digests to optimize on that you don't want to just jump in on it. AND diluting a perfectly prepared human cancer cell line digest and optimizing on that isn't something I find helpful. In fact I find the results more misleading than anything else. We didn't pick K562 as a proteome digest standard because it is difficult to work with...sort of the opposite...and no cell I've worked with has been anywhere near as convenient...)
There have been a flurry of new methods for diaPASEF recently and I probably can't get to all of them and remain employed.
I'm going to talk about regular plain old diaPASEF (first described here)
and py-DIAD (described here), but maybe more importantly Syncho-PASEF which py-DIAD enables and was described here.
and Slice-PASEF (described here)
First of all, diaPASEF is just DIA where you allow the TIMS to provide some extra level of selectivity. On the first level it really isn't all that different from using FAIMS or Selexion in conjunction with DIA. You get reduced background thanks to the IMS aspect of things so you get a better S/N ratio on your ions.
HOWEVER. That isn't all of it. While peptide m/z isn't the only factor that affects how an ion moves in collisional cross section space (CCS, which is a function of the 1/k0 [ pronounced "won over kay knot"] value the raspberry pi that powers the TIMSTOFs generate) it does play considerably into the CCS.
For example, when you look at the Mobilogram (pronounced "Mo Bill O. Graham") you see how m/z reflectes on the 1/k0 value.

One thing we need to think about is what the TIMSTOFs do quickly and what they do less quickly. If you leverage these properly you're going to get the best results and -- best I can tell, that's the main thing all of these new methods are doing.
Quads are super fast, but that speed decreases as the window they need to scan through increases in size. (Quad speed is generally described in amu/second. Which is why QQQ marketing will tell you how many SRMs you can get/second but you can only get that speed if you use a uselessly small quad isolation window and virtually no ion accumulation / dwell time.) TOF is super fast until you start averaging scans. TIMS ramping is pretty fast, depending on how you do it, but typically not as fast as either of the above. And ion accumulation is, by definition, slow and the more ions you want to accumulate the slower it is. Now what you need to do is think about leveraging these things in the most efficient ways. Some of this is actually done behind the scenes by the Raspberry Pi, something that I think the Slice-PASEF team might have found a pleasant surprise.
Back to the image above for diaPASEF.
In regular old DIA/SWATH with no PASEF we'd just accumulate from 800-850 (900 as drawn) m/z and fragment everything. But the TIMS isolation gives you something cool that you can do here on top of it. What if you took that 800-900 m/z and you did it twice, once with only the ions with 1/k0 of 1.2 - 1.1 and a second time with 1.1 - 1.0 as I've again drawn with amazing aptitude with just a mouse and my nondominant hand below?

However, the TIMS isolation and accumulation for each packet takes time, so you've got to keep track of that. Worth noting, the PA in PASEF is parallel accumulation. You actually have the capability of accumulating multiple "packets" of ions at a time. There are limits to that in regards to what ions can be accumulated at the same time and how many of them there can be. So you have to keep those limitations in mind. I won't get into that here, not because I don't understand it -- because I also don't want to.
In regular old DIA you'd just use more narrow m/z isolation windows to increase your selectivity of ions and you can do that here, but more windows equals more time for both this as well. The trick is finding the ideal compromises, but you can see how this has advantages, I hope. The default methods do seem to use 3 IMS windows to cut up each m/z DIA windows. Cool, right?
Let's get to the problems, though.
What about the ions that are outside of the normal-ish m/z 1/k0 isolation windows? Hmmm...what peptides probably behave less predictably than every other tryptic peptide....oh....the PTMs...?
Yeah. The PTMs. This is where the first py-DIAD paper steps in.
https://www.mcponline.org/article/S1535-9476(22)00087-1/fulltext

py-DIAD is an offline tool that allows you to draw your ion mobility windows in funny ways. It helps me to think that it is sort of like variable window SWATH but you also get control over your 1/k0 windows.

Now that you've got py-DIAD and you can do whatever you want with your diaPASEF windows that opens up a bunch of other possibilities. And this is where Synchro-PASEF comes in. In this, the goal is to get ULTRA SELECTIVE so that you get the vast majority of the +2 charged peptides in the cleanest way possible.
Remember what I typed above? That quads are super fast by the AMU? (P.S. Try a full scan on a QQQ. SLOOOOOW.) Also if you jump from one quad isolation window to another that also requires time. So Synchro-PASEF keeps that in mind and continuously ramps the quad. Fast by AMU, so then it cuts very small m/z /amu windows across the ion beam in a continuous order. The end result is that you scan right through the high density area where the vast majority of your +2 peptides are using tiny DIA windows. Super selectivity, by still being really fast.
Stolen from the paper. So here you've got two different experimental goals. 1) Catch everything even if it is weird (PTMs) 2) Catch all the normal things with incredibly high efficiency!

Vadim is concerned about DIA duty cycle. In his definition, a 100% duty cycle would be where every precursor is fragmented all the time. If you DIA two windows then you're at 50% duty cycle and 10 windows puts you at 10%. What he and his team tried to do is maximize duty cycle by swapping the original diaPASEF windows sideways.
Compare this supplemental figure from the Meier et al., paper supplemental with the SlicePASEF workflow

I'm not sure this was the best received article in the world and I suspect there are still people who would find a link to it annoying. The truth, though, is that if you're doing DIA you're probably doing this to some level.
To improve the selectivity of SlicePASEF you do cut up the wider windows into smaller ones, but that increases you duty cycle. Pros and cons.
Okay. This is as far as will get on this but it made it clearer to me to write it. There are more and maybe I'll get to them.
Thanks for people who were unfortunate enough to read this, I need to drop two additional links here.
midiaPASEF -- https://www.biorxiv.org/content/10.1101/2023.01.30.526204v1
SpeedyPASEF -- https://www.biorxiv.org/content/10.1101/2023.02.17.528968v1

If you promise not to break the server, I'll give you the link here!
WOOOOOOO! I've been dreaming about a program like this for years and I bet you have too.
I don't know if there is a paper yet. I clicked the wrong link and saw that this had moved from "coming soon" to live. And the manual references a paper (yo, if you need a reviewer, I'm only at 75 for the year so far)
Why am I super stoked about this one? Check out this option!!

I've rambled about this before, but for just about everything I do the phosphorylation information exists in two possibilities.
1) Protein level increases (but phospho-level remains the same). If you just phospho- enrich, who knows which one it is? Not you, that's for sure.
2) Protein level remains static but phosphorylation site occupancy increases.
I don't know why exactly, but to most people I know the amount of ERK vs phospho-ERK is a very important thing. So important, in fact, that they'll do western blots.

I'm on my second read through of this new study in JPR. After the first one I realized my coffee was sitting on the counter untouched. Having rectified that, it is starting to make sense. Why would I read it again? Because I wouldn't mind having like 50% more confident phosphopeptides and -- like you, I bet -- I've always had some skepticism when I see at the PSM level multiple phosphosites observed that I don't see in my final peptide/protein report.

I just tried and realized that I don't have the time to do this justice, and it is better described in the apper -- and I should be getting ready for my daily death-defying commute through the Thunderdome.
The important thing here is that 1) it doesn't look like we are doing it right (taking just the best scored PSM for each potential PTM site and rolling that up) 2) it doesn't look that hard to implement what this group did and 3) this is very convincing that we're both losing information AND it this is one method that might allow us to get it back. Super cool study and worth at least one readthrough.

Did you know it used to be really common in qPCR to never use the wells on the edges? I think it is still recommended for the old thermocycler that I have.
Now that a lot of us are moving to plates for proteomics and smaller and smaller volumes it becomes a much bigger deal, I guess.

Due to peer pressure I'm doing a bunch of LFQ diaPASEF based single cell proteomics and since I'm only loading 2 uL in each well of a plate I run the edges first if the gradient is short or skip the edges entirely if the gradient is long. If a darned well is going to dry out it is almost always going to be the edge.
And, BTW, fuck these stupid silicon plate lids. They suck. You're way way better off using a tape based sealant for your plates and then just setting yourself up with one sample per well (since you've punched a big hole in the top and it will evaporate

I've heard concerns about the glue on them, but we're several thousand runs in with this one https://www.thermofisher.com/order/catalog/product/60180-M143 (M143) and haven't seen any issues (DISCLAIMERS OVER THERE! -->)
Disclaimers over there --> this just appeared to work for us. The same big batch of files that were taking forever finished overnight after I made these changes. Disclaimers -->
This is going to be very quick and largely unscientific, but if you're facing huge SpectroNaut bottlenecks like we have been, all of this has helped.
1) Run everything to and from a high speed solid state drive (especially the Temp folder)

According to this computer nerd page, our NAS drives read/write at less than 200 MB/s


2) We put in more (and faster RAM)
Our PC had 64GB of RAM that was rated at 2.7 whatever units. For $700 we were able to take that out and put in 128GB of RAM that was rated at 3.7 whatever units. 1.6x faster? Maybe?
3) Turned up the fans on our processing PC and moved the settings from "Quiet" to "High Performance"
This box has an AMD processor and I found some software from the vendor that allowed me to see what was going on with all the PC things. There were settings on the water cooler, etc., where I could choose "quiet", "balanced", or "high performance". Obviously I put it on the last one. AND it allowed me to change the lights on the PC. Since we know that PCs need to be cooler, I set all the lights inside the PC to blue. The PC is now louder and, at least at a psychological level, it appears to be much faster.
4) On big queues >100 files, I've also found that converting the data to HTRMS first appears to help. There is a little utility with SpectroNaut that can do this, however, if your 100 files are 400GB, you may now have 800GB of data, but these HTRMs things do appear to process faster (thanks Dr. Neely for this tip).

If you're cool I'll probably just agree to do stuff for you without inquiring about any of the details.
So when one of the best technical mass spectrometrists in the world (Bogdan!) asked me to talk at some thing in May, I just said yes and put on my calendar that I was flying to Boston today.
No flight for me today! I'm speaking remotely at this thing!

2) Wooo! Wide Windows? My kid carries a jackalope with him to preschool whenever he isn't too sick to go. 2 year olds are germ factories of despair.
3) Meh. I've seen that one.
4) YEAH. If you don't know Claudia she's an absolute force of technical capabilities.
5) I just had to have a single cell poster beside Dr. Parker. People sort of spilled over from hers to talk to me. She's got some stuff in works that should be out any time now that is just ridiculous.