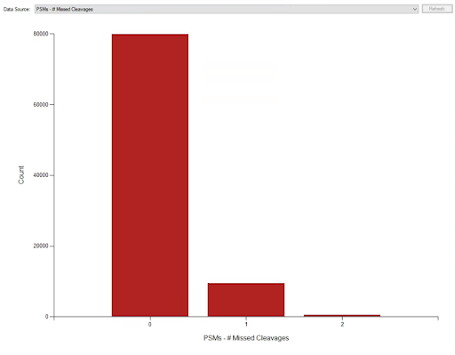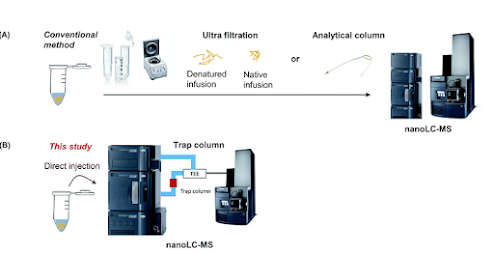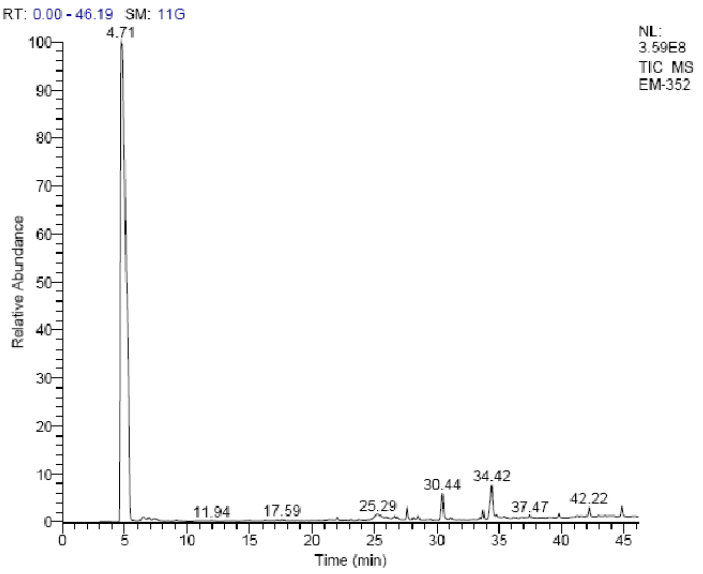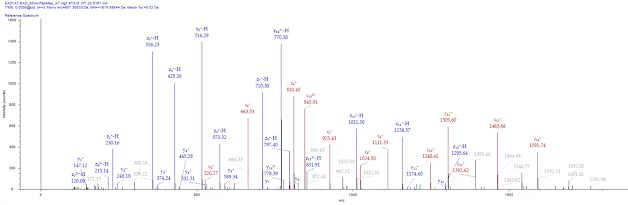
The image quality above is not this blog input's fault. This is actually what it looks like through the publisher's online browser that they keep forcing us to use for some reason.
HOWEVER. This paper is 100% recommended for absolutely everyone. This has the answers to the big questions that I'll call friends who work in core labs to ask.
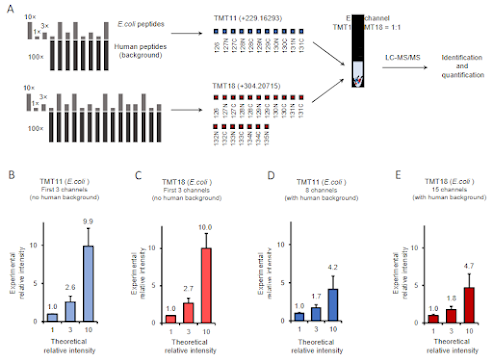
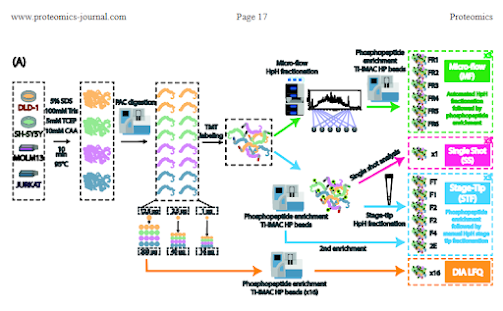
Multiplexed phosphoproteomics -- when do I label vs. when do I enrich? You can enrich then label then mix, and you can label and mix then enrich. I've done it both ways out of necessity, but what should you actually do?
TAAADAAAAA! This new permanently on my desktop forever paper asks the hard questions and answers them. The authors obviously knew how important these questions were, and had no question whatsoever that this was going to get published, so they drew the figures in crayon just for fun.

(I realize it is the conversion process into the publisher's super annoying interface, but this IS the version that I will keep a PDF of, and not only so I don't have to see the coffee cup filling icon on my screen. I happen to like the crayon effect.)