diaPASEF has been all the rage recently based on really amazing levels of coverage that we've been seeing on both long and short gradients at every meeting (and an increasing number of new papers).
As an operator of a TIMSTOF who might want to start exploring these relatively new superpowers, here is my advice from a focused holiday weekend of getting this all up and running.
Let's get this out of the way first. Newer versions of TIMSControl (released at Halloween ASMS 2021) have the ability to create windows based off of your DDA data.
NO. MATTER. WHAT. DO. NOT. USE. THIS. FEATURE.
For real.
If you do use that feature, you'll get the stupidest DIA windows that ever happened and they use up huge amounts of your cycle time. Real examples!
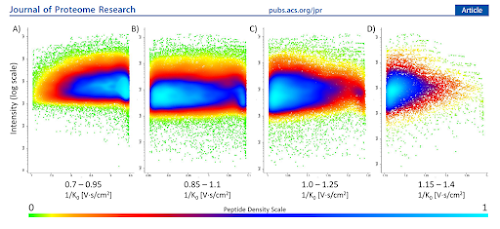
Now...if you are interested in the peptides that have an m/z of 100-150 and the extremely selective ion mobility isolation range of 0.6-0.95, please ignore what I just typed. I do think that it is possible to see doubly charged dipeptides and some smaller tripeptides in this range. Don't quote me, but I do think that this window would allow you to see such interesting targets such as RK and RAK and RGK, and if that's your thing, ignore me and let TIMSControl build your windows.
What is also important here is that : If you did import your data from DDA, you can not under any circumstances whatsoever, delete or alter those windows. They are stuck in that method forever until the end of time. Trying to alter or delete those windows will put you into a perpetual loop where TIMSControl will keep reloading that DDA file (which will take several minutes per cycle) and it happily do this forever. Your best best bet is to....if you have a TIMMY...you already know what I'm going to say...
It's time to fire up:
-the most stable
-best written
and by far, the
- most utilized executable in the history of this vendor's mass spectrometry instrumentation...
The STOP ABSOLUTELY EVERYTHING button. (Sometimes people think this is funny that this exists, and considering the absolutely amazing number of steps needed to switch from trapping to nontrapping on a NEO attached to a Fusion 2...maybe other instruments would also benefit from this button).
Now that we've got what to never ever do out of the way. What should you do? Well, you should manually type in your windows and you should use big windows for short gradients and small windows for longer gradients. It looks like it's best to just manually type everything from someone else's paper and just use their gradient conditions exactly.
Want a step by step guide? This one is great!
Another caution here. It is possible to pull the instrument method out of the .d data files using Data Analysis. I haven't had much luck with these, particularly from studies out of some place in Munich. My guess is that the software that they work off of is a version or ten ahead of the commercial releases. Also, my Flex doesn't respond well to Pro files all the time, so that might be a Flex specific issue.
Aha. There it is! I've had this open on tab 84 on my iPhone SE that is perpetually hot for some reason. This is brand new and from the title you wouldn't guess that it is a thorough diaPASEF optimization study (for plasma).
Their optimization shows that they get much better data tweaking things specifically for plasma because it isn't as convenient as HeLa.
This is obviously a very new toolset and things are developing very quickly and I may have a bug and might just be a jerk. The data you get when using diaPASEF correctly does appear to be great. Honestly, even when I wasted cycle time with stupid windows, my diaPASEF runs still outperformed a QE Classic running clever variable DIA windows from a new preprint out of Slavov lab (n=84, processed with DIA-NN, allowing it to make the library and pick the fragment ion tolerances.)
However, you probably didn't remove ceiling tiles to get slightly better data than a ten year old instrument. Since I threw out singly charged molecules in DIA-NN this was probably unfair to the TIMSTOF and all of the dipeptides it spent time analyzing over the holiday weekend before I put an SOS out to the totally awesome Dr. Florian Meier who helped get me back on track.