The last one was well received! How 'bout one that took us months to put together?
For more details, check this out:

1) You need to have a low mass fragmentation scan and a high mass fragmentation scan. This is due to TOF effects through the quad prior to pulse activation that sends the fragments into the TOF. Oddly, enough someone solved this problem recently, but I don't think they'll share it with this vendor.
End result is that your (theoretical) 120Hz instrument is going to be a 60Hz instrument. The cool part is that since you have two separate packets of fragments you can do different things with them. The obvious idea is to use a relatively low energy on the packet you'll use for sequencing your peptide and get stupid with the fragmentation energy you'll use for the packet for reporter ion release.
2) While you can do TMT18-plex quan if you want (see supplemental of preprint) it might not be completely ready yet. Remember how when the TMT10plex came out and all of us with OrbiXL/Velos systems used 35,000 MS2 (4 scans/second or something? 3.5?) and it worked okay, but anyone with the Elite system made fun of us because they got baseline separation at 60k, but no one stopped to check to see if there was any biologically relevant difference because we didn't know how but we didn't want to be made fun of by labs with vendor deals or infinite research funding so we basically stopped? Same principle here. A little worse because you can't centroid the TIMSTOF data perfectly yet. At least 2 groups are working on it. I'm eagerly awaiting final products.
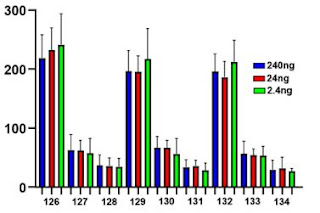
I recommend using the TMTPro18 reagent and then using every other tag. We use the n tags for consistency and keep the c tags for making spectral libraries and other one-off type experiments. With the TMTPro18 you can 10-plex this way! That's the best you could do with anything just a couple of years ago!
3) The mass accuracy isn't going to be as good. OR IS IT? I'm super proud of this idea because I don't think anyone thought of it first somehow and I was like "I'll find out later someone did this in 2005 and I should have known" but -- check this out. In a TMT quan experiment you always have a reporter ion. I'm doing SCoPE-MS so my carrier channel is always this high abundance flag in every spectrum. I know the exact mass of that! Why don't I just recalibrate each file on that fragment ion? Taadaa!
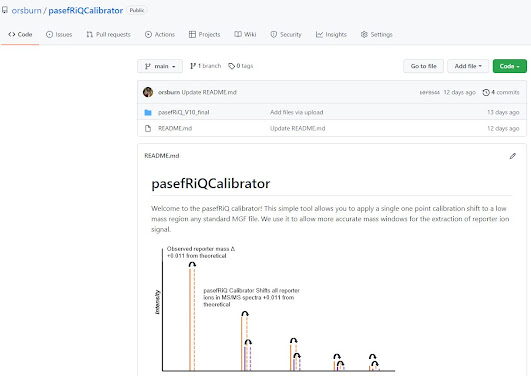
I do also recommend doing the main mass calibration with a syringe using the Agilent low concentration TOF mix because it has low mass ions, prior to starting any TMT/iTRAQ batch, because if you calibrate off of the filter with your lowest mass being 622, you can get wobbly in the low mass range. This can still adjust it, but it's better to start close.
4) Adjust your TIMS to match your eluting ions and keep in mind that TMT6/10/11 and TMTPro migrate differently to each other and unlabeled peptides. If you want the best coisolation interference, use a really narrow range. 0.8-1.3 can work really well. However, if coisolation isn't the biggest concern for your sample (like there is very little around that could coisolate in your sample, crank that number up).
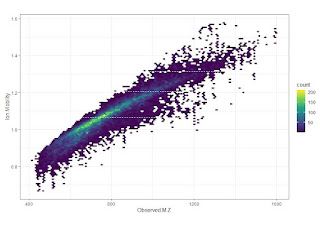
TMT11plex shown, this didn't make the paper, butI love these hexagonal R plots! Almost all of the ions are between 0.8 and 1.4, literally ever identified peptide is between 0.7 and 1.5, so even narrowing it down a little will increase you ion mobility resolution for each MS1 and MS2 spectrum.
5) Note on quad isolation -- the vendor default TMT template (which I am lobbying to be renamed pasefRiQ because that's just cooler and less concerns of lawsuits!) uses a 2Da low mass and 3 Da high mass cutoff. That works for signal intensity (you didn't buy this thing because of it's great quad...I hope....?). Using the TMT TKO standard 1.5 up/1.5 down seems to be the best in my hands, but I didn't try 1.2 or 1.26, so this isn't super extensive. You hopefully bought this big thing because you thought that 200 resolution in ion mobility was probably an exaggeration, but it would be killer if it wasn't.
What else? Oh yeah! How to process the data? That's easy. Use basically anything you want.
Things that can do quan on MGF spectra?
Proteome Discoverer (including the free one).
SearchGUI + PeptideShaker + Reporter (this is still in beta).
Fragpipe will complain about it with popups, but I think you can just lie to it, say this isn't ion mobility, what are you talking about? it's just an MGF and you're fine. Can't say with 100% certainty, though.
A couple steps are necessary for Proteome Discoverer to process right.
You do need to use your spectrum selector so it knows what these files are. I suggest you lie and say the files are FTICR. This helps you keep all of your decimals places.
Tell it that unrecognized fragmentations are CID (honesty this fragmentation looks more like HCD and the two things are basically the same, so do what you want here) and give it your MS1/MS2 resolutions.
Big thing here if you are using MSAmanda, go under advanced in the node, scroll allllll the way to the bottom and max out your number of MS2 spectra per search (50k) you might have 200k MS2 per file. At 10k a piece that's a lot of searches. I recommend percolating. I also highly recommend using the most specific database that you can. It isn't a stretch to say that you might be giving Percolator e7 spectra to look at. If this is is RBCs do you really need all those mitochondrial peptides for all your tools to fuss over?
Recommended mass accuracy? -- If you've recalibrated your MGFs with MSFragger (crap. maybe I should write up a real tutorial. More later maybe, but MSFragger can recalibrate your spectra in the high mass range and it's AMAZING) -- 15ppm MS1 and 25ppm MS2/0.02 Da will work great. If you haven't calibrated I like 30ppm MS1 and 0.05 Da MS2. If you haven't calibrated in a month...?...go calibrate it.
Quan on one hit wonders might be wonkier than usual, but if you look close you'll see it probably isn't quan. It is that your FDR filters can let through a few more bad hits than you're used to. More on this later, but spectral libraries are AWESOME for TIMSTOF data. With the multithreaded MSPepSearch you can knock out a TIMSTOF MGF in like 90 seconds, no problem, but I think that the Orbitrap generated spectral libraries might not be as awesome as ones developed from TIMSTOF data.

No comments:
Post a Comment