
I looked for some of my LTQ files from my postdoc to use as an example, found that some of these old drives might not work anymore (gasp! what will I ever do without 4 pounds blocks of steel holding low resolution files?) and then poked around on PRIDE until I found a few published studies with chromatography like this one above.
Just to be clear, my chromatography looked like this a lot when I was moving from glycans (my grad work) to proteomics and I've seen lots of other people, primarily people used to analytical chromatography, end up with proteomic files that look like this. It's also very often an issue when people are getting used to PepMap. If you think this is your file, it probably isn't. I made chromatography suggestions on multiple papers I reviewed last year where I suggested making some alterations to chromatography for their next study. It'll take you 10 min on ProteomeXchange (assuming you can download files on a 10GB connection) to find several nice examples, but I'm going to pick on this random one.
As fast and powerful as today's instruments are, you can get data out of files like this, but some chromatography optimization can get you much further.
For some hypothetical numbers, let's drop this file into RawBeans to see how things look. 
Holy cow. This instrument is FAST and I'd guess that getting the maximum number of MS2 scans was a primary goal of this file because it looks like there were in excess of 60 MS2 scans allowed between each MS1 scan! Even if that was an exceptionally rare occurrence, the apex of the chromatography is around 30 minutes and we see that here and there the instrument was hitting 40 MS2 scans for every MS1 scan. However, if you look across the board it looks like the average was probably a whole lot closer to 15.
That math kind of makes sense because RawBeans says:

So, 61,272/100min/60seconds gives around 10.2 MS2 scans/second. If we look at the area from 0-17 min where there is nothing and the space around 95 minutes, the active gradient was probably closer to 15/second. Honestly, not that bad, but again -- that is an extremely fast instrument.
I dug a little and found a RAW file around the same length with a more flattened (PepMap) chromatography gradient --

RawBeans says --
Realistically, not an improvement, but check this out --

This instrument isn't anywhere near as fast as the one above. The maximum number of MS2 scans that ever occur are around 15.
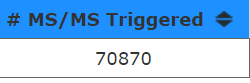
But, 70,870/130min/60seconds = is around 9.1 scans/second. That makes this density plot look maybe just a little misleading? I think that the pixels size used to indicated each number of scan events must just max completely out on this scaling.
Either way, we've got instrument A that is capable of 60 MS2 scans/MS1 and instrument 2 that maxes out at 15 MS2 scans/MS1 -- and because run #2 flattened out the PepMap mountain the much slower instrument gets comparable numbers of scans.
Here is the gradient, as saved in the second RAW file

File 1? Well, it gets to 45% Buffer B in 80 minutes. Which honestly makes sense for analytical flow C-18 for a lot of organic molecules (HyperSIL Gold, for example, maybe you need 45% B to push your molecules off), but if you think about where 20% or 25% B would occur on file 1:

Now, what's funny about this is that if you just extract a couple of peaks from these 2 files, on the surface they look pretty good. In fact, the PepMap Mountain file looks like it has sharper overall peak shapes, with a full width half maximum (estimate about 50% of the peak height and then figure out how many seconds it is across that, or FWHM) that is better than the other file.
While FWHM is a valuable metric it is NOT the most important one when thinking about mass spectrometry cycle time. What you actually care about in LCMS proteomics, almost always, is the peak width at the threshold where you would actually trigger an MS/MS scan.
When you look at the PepMap Mountain file in that way, this is where you see the problem.
This is actually tough to visualize, but what I did here was extract a randomly selected precursor, extracted it at 5ppm, and used "point to stick" to show it's occurrence. (Then I blocked out anything that might be personally identifiable in this file in red, I'm not trying to pick on anyone.)


The imporant part here is that this peak, when allowing a signal of 50,000 counts to trigger, was triggered 6 times and that's why -- long long story even longer -- the percentage of MS2 scans that are converted to peptide spectral matches is significantly lower for the much much faster instrument with the PepMap Mountain chromatography compared to the significantly slower and older system.
I guess I didn't mention that part. And the whole reason for this post!
Here is the scenario that started this! Brand new system A running a 2 hour gradient was compared to 9 year old sytem B running a 2 hour gradient. 9 year old system got 2x more peptides. Number of scans per file looked about the same. I can't share the actual files, but it only took about 4 minutes of searching to find published data to illustrate exactly what happened and for some reason I spent an hour making screenshots and typing when I should have been sleeping, but in my defense, I didn't feel awake enough to work on things that I get paid to work on.
I pick on PepMap, but only because it is where this is the most extreme. Compared to any other C-18 I've ever used, stuff comes off it the earliest. I've often wondered how much a misunderstanding of this property leads to it's lack of popularity, but even at 100% aqueous you do seem to lose more phosphopeptides with it than with anything else and I'm pretty sure that's why CPTAC stopped using it.
I'm going to stop, but here are some related past posts:
Even more boring chromatography post!
Crap. This one is even older. I wouldn't even post this, but I did review an LTQ Orbitrap study recently where unit resolution dynamic exclusion was used. People I work with today were in middle school when I wrote this, but I commonly wear shoes to lab that are older than some of them, so that's not all that weird, I guess. OH! And it has a great example of where my chromatography was a mountain! Totally worth linking here.
No comments:
Post a Comment