
Everybody knows someone who has or is getting RNA-Seq data. And we've seen the awesome papers where using this data has given us new mutations and cleavage sites and things that we didn't get from a normal FASTA. Connecting those two, however, still seems to require a dedicated bioinformatician.
From my perspective I figure the easiest way to get these things set up is to change the RNA-Seq data into a FASTA that I can input into my normal proteomics pipelines (cause I already know how to use those).
Even this, however, isn't without problems. If you take the nucleotide sequences as they are you still have to change those into proteins. The simplest way to do that is to do a 6 frame translation.
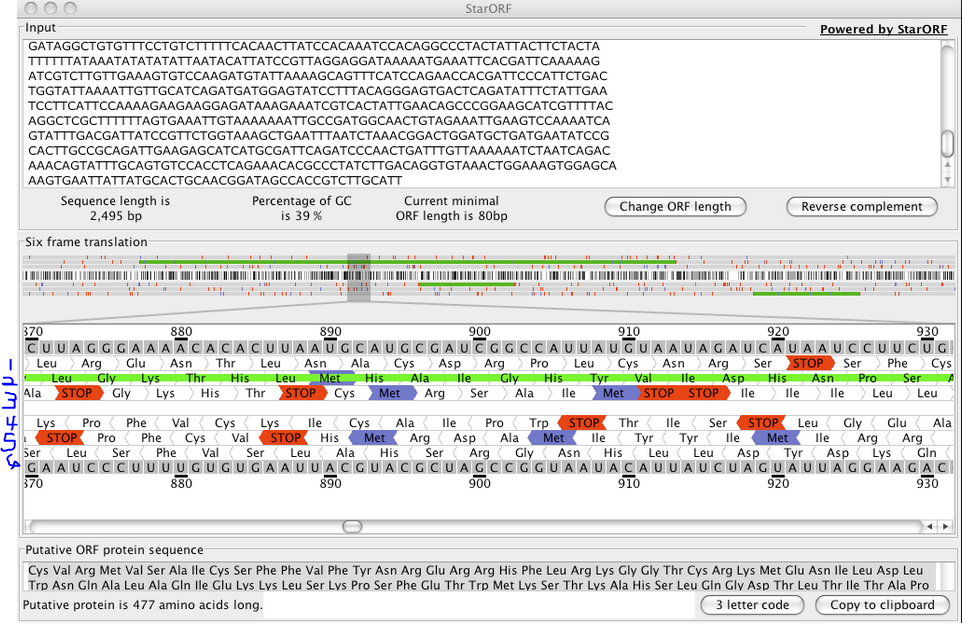
Check out this example. We line up the nucleotides and pick 6 different starting points (here counting both DNA strands). We know that 3 nucleotides code together for one amino acid, but we don't know what our frame of reference is. Where do we start counting? In a 6 frame translation you go ahead and do them all.
In this nice example above, four of the possibilities can be pretty much ruled out because there are so many stop codons that they likely don't code for anything that we can sequence in a shotgun experiment. But ruling these out can get real tiresome real fast and a LOT of sequences aren't this clear cut. So you basically make a database that is 6x bigger than it needs to be with 5 sequences that aren't real (lets not even consider what this does to FDR. Lets not. Its late. And we aren't even talking about introns and exons, which is worse).
Franziska Zickmann and Bernhard Y. Renard think we should be smarter than this. And they have a solution that is better that they detail in this open access paper that they call MSProGene.
This whole packaged solution isn't exactly what I'm still wishing for (easy, intelligent FASTA generation with redundancy removal), but it shows how we can use the sequencing data to help us with another big problem -- protein inference! We can combine what we know from the genes present to help us figure out what proteins our peptides came from! MSProGene is a complete package that does the work from RNA-Seq and LC-MS data all the way to peptide and protein lists (its uses the MS-GF+ search engine).
While its still probably a tool aimed for bioinformaticians to work through combining these rich and complicated data types, it undeniably adds some great new tools and insight into this pipeline.
No comments:
Post a Comment