
A smarter person in a very competitive niche would probably blog about a lot less of the cool things that he/she reads, but we ain't known for smart in Tallmansville WV. We're mostly known for having really extremely long grudges that defy logic....and...mining catastrophes....
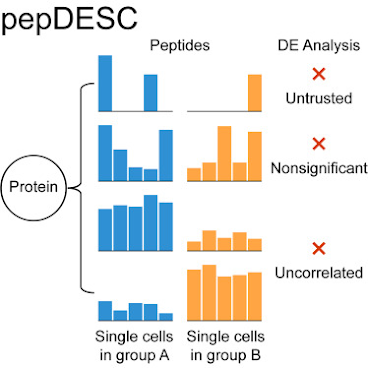
PepDesc goes at SCP data significance from the peptide level with really clear logic and through an interface that is surprisingly less than miserable to set up.
I will give you these corrections, however, if you're interested.
The correct installation with devtools is not the one on the Github (on Windows with R 4.2.0)
It is "devtools::install_github('dionezhang/pepdesc')"
and "load" has no function, run like normal as
library(pepDESC)
My only complaint is that your peptide level data needs to be loaded as a *.rda, but otherwise it is incredibly simple.
As an aside, I did hit a maximum matrix in Proteome Discoverer for single cell data. At >1,500 cells I broke the visualization for peptide groups in PD 2.4SP1. I've got PD 3.0 now, so I do have to get some data out the door before I start the full migration to the new software.
Super impressive work from a solo(!!) author I don't know(?), who appears to have ran single cell label free proteomics on 60 or so cells to support the development of this code(?) Badassery.
No comments:
Post a Comment